弥漫大B细胞淋巴瘤(DLBCL)是最常见的非霍奇金淋巴瘤类型,其中活化B细胞样亚型(ABC-DLBCL)因预后差、易复发而长期困扰临床。该亚型常伴有CARMA1-BCL10-MALT1(CBM)信号体的异常激活,导致NF-κB通路持续驱动肿瘤细胞增殖与存活。尽管已有针对MALT1蛋白酶活性的抑制剂进入临床试验,但其仅能部分阻断信号传导,且可能引发免疫失衡相关的炎症毒性。因此,开发更全面、更安全的靶向策略成为研究焦点。
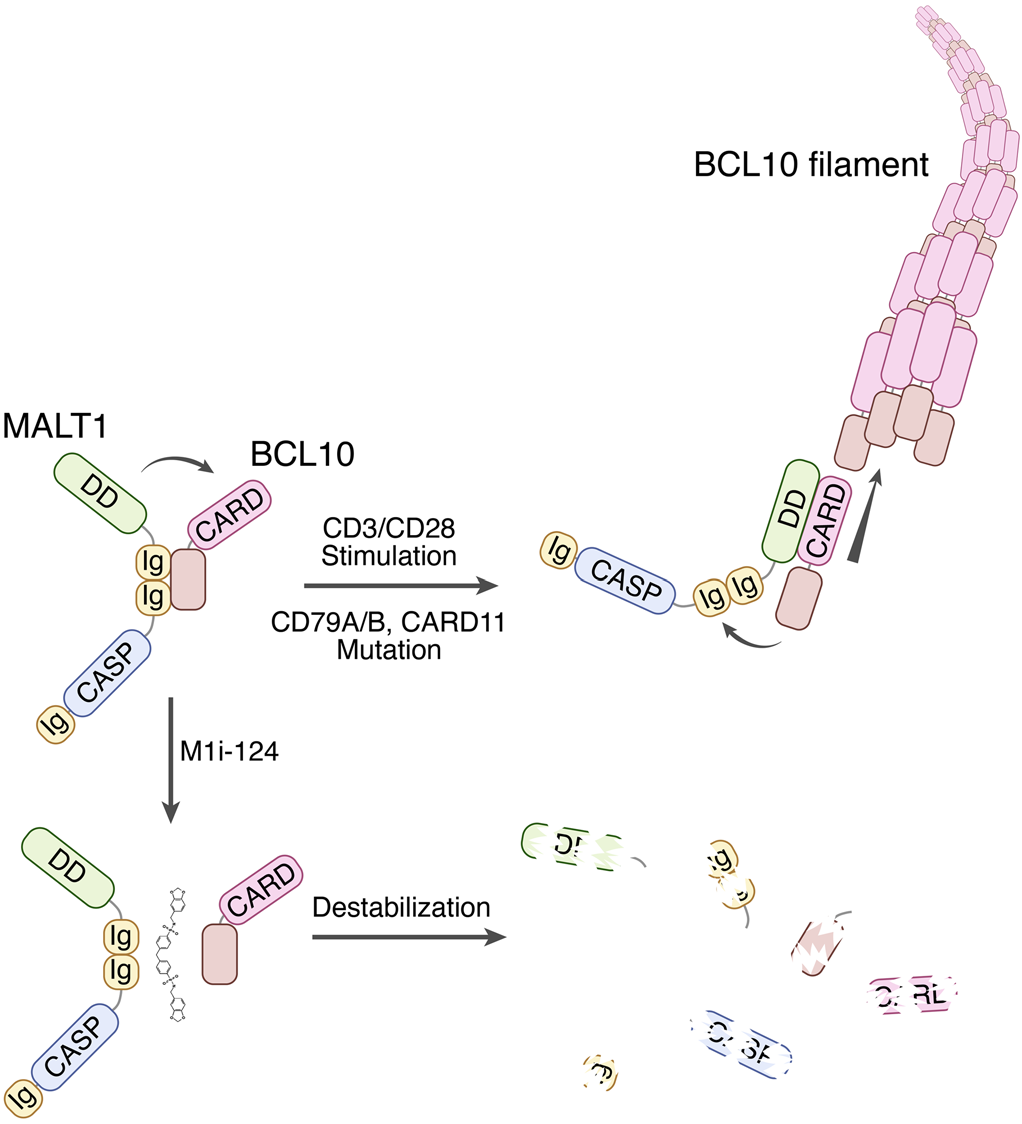
近期发表于《Journal of Clinical Investigation》的一项研究1揭示了一种全新的治疗思路,通过小分子化合物直接干扰BCL10与MALT1之间的蛋白-蛋白相互作用(PPI),从而从根本上破坏CBM信号体的组装。研究人员利用结构生物学指导的虚拟筛选,成功鉴定出首个靶向该互作界面的小分子抑制剂M1i-124。该化合物不仅能有效抑制MALT1的双重功能(支架与蛋白酶),还能诱导BCL10和MALT1蛋白降解,在ABC-DLBCL模型中展现出强效且特异的抗肿瘤活性。这一发现不仅为难治性DLBCL提供了潜在的新药候选,也为其他依赖CBM信号的恶性肿瘤及自身免疫病开辟了新的干预路径。CBM信号体是B细胞受体激活后触发NF-κB信号的核心复合物,其中MALT1作为效应蛋白,通过与BCL10的相互作用发挥支架和蛋白酶双重功能。传统观点认为MALT1的死亡结构域(DD)与BCL10的CARD结构域结合,是两者的主要相互作用方式,但最新研究表明,在未刺激状态下,MALT1的两个串联Ig样结构域(Ig1-2)与BCL10之间存在高亲和力结合,提示这一区域可能是调控信号起始的关键节点。研究人员通过共免疫沉淀(co-IP)实验证实,仅包含Ig1-2结构域的MALT1片段即可高效结合BCL10,而单独的DD片段则几乎无结合能力。进一步分析显示,MALT1(Ig1-2)与BCL10的解离常数(KD)低至29 nM,表明其具有极高的结合亲和力。通过对晶体结构(PDB ID: 3K0W)的分析,研究团队识别出位于Ig1与Ig2交界处的一个深疏水沟槽,多个突变实验验证该区域氨基酸残基(如V189R、A203-A205、A257-A259)对BCL10结合至关重要。这一发现首次明确了MALT1上一个可成药的蛋白相互作用界面,为后续小分子抑制剂的设计提供了精确靶点。拓展阅读
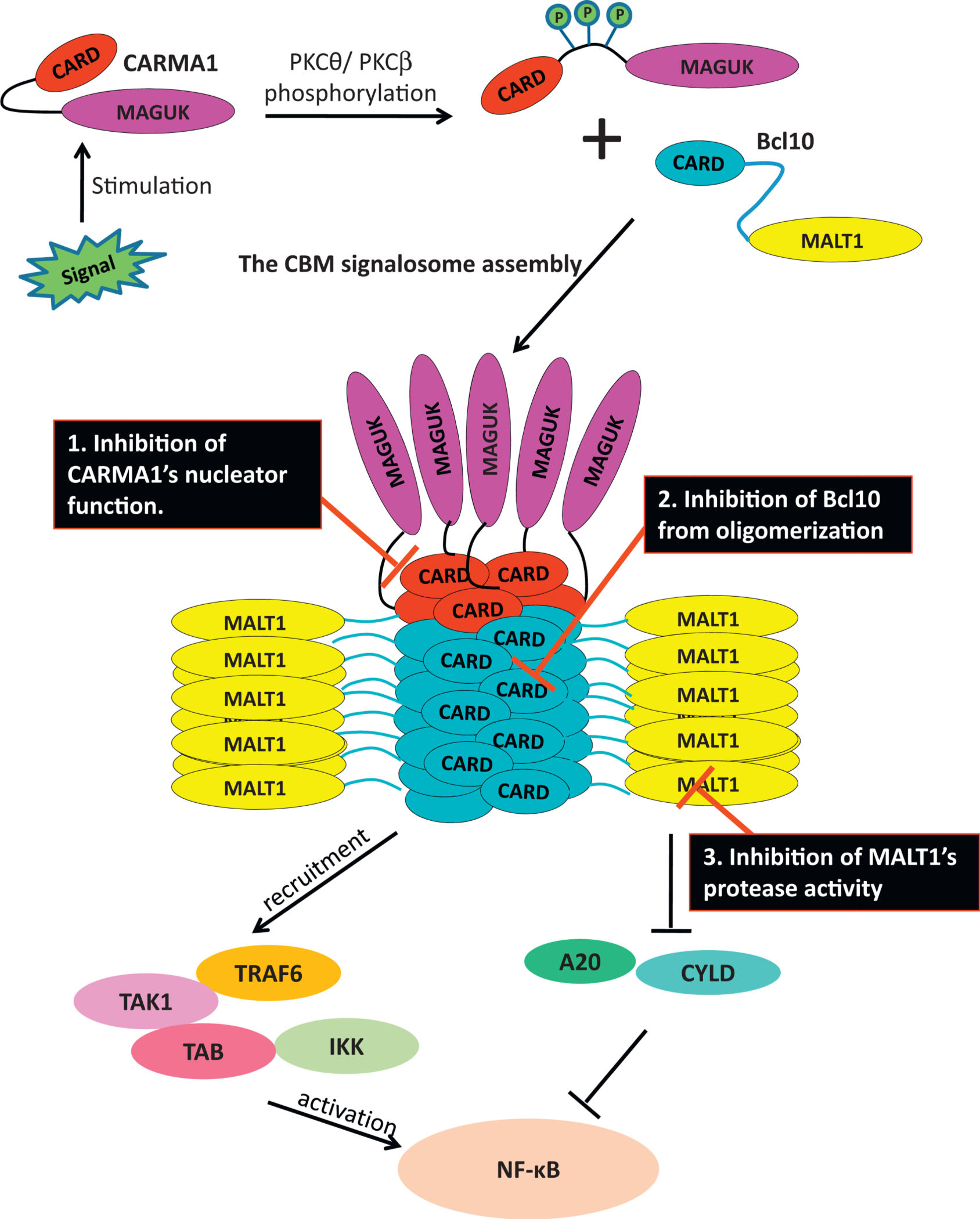
CBM信号体(CARMA1-Bcl10-MALT1复合物)的异常激活是淋巴瘤发生发展的关键驱动因素,核心机制为NF-κB信号通路持续失控。在弥漫大B细胞淋巴瘤(DLBCL)的活化B细胞样亚型(ABC-DLBCL)中,CARMA1卷曲螺旋区常出现功能获得性突变,可解除其自抑制构象,无需PKCθ/PKCβ磷酸化即可自发寡聚化,进而持续驱动Bcl10丝状组装与MALT1激活,形成稳定CBM复合物并激活NF-κB。
在黏膜相关淋巴组织淋巴瘤(MALT淋巴瘤)中,染色体易位是主要异常形式,t(11;18)(q21:q21)产生的cIAP2-MALT1融合蛋白,其BIR结构域可增强MALT1蛋白酶活性,持续切割A20、RelB等NF-κB负调控因子,同时维持CBM复合物支架功能以招募TRAF6、IKK复合物,强化NF-κB信号;t(14;18)与t(1;14)易位则使MALT1、Bcl10基因紧邻Ig重链位点,导致二者过度表达,加速CBM复合物组装与NF-κB激活。
持续激活的NF-κB信号会异常调控淋巴细胞增殖、存活相关靶基因,促使其摆脱正常生长限制形成恶性淋巴瘤,而ABC-DLBCL对CBM介导的NF-κB信号高度依赖,印证了CBM信号体异常在淋巴瘤发病中的核心作用2。
图1:CBM信号体组装模型及潜在的抑制方法
M1i-124:首个靶向BCL10-MALT1 PPI的基于上述结构信息,研究团队采用Discovery Studio 3.5平台中的LibDock模块,对ChemDiv数据库中300万种化合物进行虚拟筛选,并结合Lipinski五规则过滤,最终获得9个候选分子,其中7个可商业化获取。在这些候选物中,仅M1i-124在TMD8细胞(一种MALT1依赖型ABC-DLBCL细胞系)中显著诱导凋亡并降低细胞活力。表面等离子共振(SPR)实验确认M1i-124能与全长MALT1结合,KD值为2.96×10-4 M,证明其具备直接靶向能力。更重要的是,ELISA结果显示M1i-124及其类似物M1i-124d1可剂量依赖性地抑制MALT1与BCL10的结合,IC50分别为16 μM和34 μM,而对照化合物C741-0547则无此效应。此外,M1i-124不影响MyoD与Id1的结合,表明其作用具有特异性。这些数据共同确立了M1i-124为首个被报道的BCL10-MALT1蛋白相互作用抑制剂,代表了一类全新的精准治疗策略。图2:BCL10-MALT1相互作用模型以及M1i-124对CBM组装过程中的破坏作用M1i-124双重抑制MALT1功能并诱导蛋白降解M1i-124的作用机制远不止于阻断蛋白相互作用。在Jurkat T细胞中,该化合物可显著抑制抗CD3/CD28刺激诱导的RelB和N4BP1切割,效果与已知MALT1蛋白酶抑制剂哌卡嗪相当,同时还能减少IKKα/β磷酸化,说明其同步抑制了MALT1的蛋白酶和支架功能。在ABC-DLBCL细胞系TMD8和OCI-Ly3中,M1i-124同样有效抑制RelB切割和IκB磷酸化,并显著降低IL-6和IL-10的mRNA表达及分泌水平,这两者均为NF-κB下游促生存因子。尤为引人注目的是,M1i-124处理72小时后,可导致BCL10和MALT1蛋白水平显著下降,而对照化合物或哌卡嗪均无此效应。这种蛋白丢失呈时间与剂量依赖性,且不伴随mRNA下调,反而出现反馈性上调,提示其机制源于蛋白稳定性破坏而非转录抑制。由于BCL10与MALT1互为稳定伴侣,一旦相互作用被阻断,二者均易被泛素-蛋白酶体系统降解。这一“间接降解”效应解释了为何细胞内功能抑制的IC50(0.5–1.3 μM)远低于体外PPI抑制的IC50(16 μM),极大增强了其生物活性。本研究成功鉴定了首个靶向BCL10-MALT1蛋白相互作用的小分子抑制剂M1i-124,突破了以往仅针对MALT1蛋白酶活性的局限,实现了对CBM信号体更彻底的抑制。其独特之处在于不仅能阻断MALT1的双重功能,还可诱导关键蛋白降解,从而在ABC-DLBCL模型中实现强效且特异的抗肿瘤效果。动物实验显示M1i-124具有良好药代动力学特征和安全性,未见明显器官毒性或免疫紊乱,提示其可能规避现有MALT1蛋白酶抑制剂所面临的自身免疫风险。然而,该研究仍存在一定局限:目前尚无关于M1i-124在人体内的药动学数据;其口服生物利用度有待提升(分子量偏大、TPSA较高);且尚未明确其是否适用于携带特定突变(如BCL10截短)的患者群体。展望未来,随着更多结构导向的PPI抑制剂涌现,我们有理由相信,这类“源头阻断”策略将在精准血液肿瘤治疗中扮演越来越重要的角色,甚至拓展至自身免疫性疾病领域。参考文献
1.Kang H, et al.J Clin Invest. 2025;135(8):e164573. Published 2025 Apr 15.
2.Yang C, et al. Cytokine Growth Factor Rev. 2014;25(2):175-183.



 歆语健康发布于 3周前
歆语健康发布于 3周前 收藏专家
收藏专家