


 歆语健康发布于 3周前
歆语健康发布于 3周前 收藏专家
收藏专家外周T细胞淋巴瘤(PTCL)是一类高度异质、侵袭性强且预后极差的非霍奇金淋巴瘤。尽管近年来免疫化疗与细胞治疗取得进展,但其生存率仍低于30%,亟需发现新的分子靶点以改善临床疗效。铁死亡(Ferroptosis)作为一种铁依赖性的程序性细胞死亡形式,近年来在肿瘤治疗中展现出巨大潜力,尤其在克服耐药方面表现突出。然而,其在PTCL中的调控网络尚不清晰。
近期发表于《British Journal of Cancer》的一项研究1首次系统揭示了前列腺素D2合成酶(PTGDS)在PTCL中的关键作用。该研究不仅证实高表达PTGDS与患者不良预后显著相关,更重要的是,通过体内外实验阐明了靶向PTGDS可通过调控HMOX1介导的血红素降解和铁蛋白自噬,促进铁积累并诱导铁死亡,从而增强对索拉非尼等药物的敏感性。这一发现为PTCL的精准治疗提供了全新的理论依据和潜在联合策略,标志着铁死亡通路在PTCL中的转化研究迈出关键一步。
靶向PTGDS抑制PTCL进展并促进铁死亡过程
为探究PTGDS的功能,研究人员采用慢病毒转染技术构建PTGDS敲低和过表达细胞系,并使用特异性抑制剂AT56进行干预。结果显示,PTGDS敲低或AT56处理均可显著抑制PTCL细胞增殖、活力、侵袭能力,并诱导G0/G1期细胞周期阻滞及凋亡增加。体内小鼠移植瘤模型也验证了这一效应,敲低PTGDS组或AT56治疗组的小鼠肿瘤生长速度、体积和重量均下降,活体成像显示生物发光信号减弱。
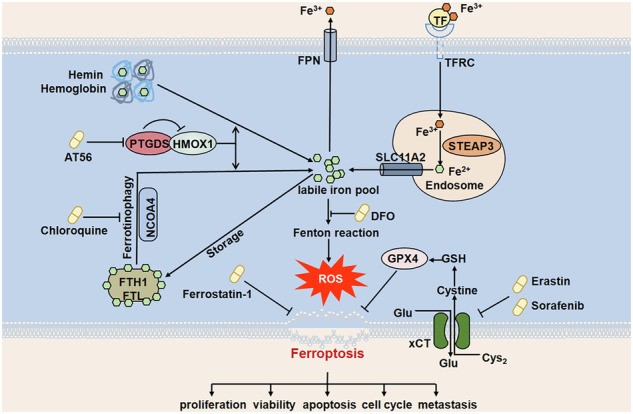
该研究最核心的机制突破在于揭示了PTGDS与血红素加氧酶1(HMOX1)之间的直接相互作用。分子对接、免疫共沉淀(Co-IP)及共聚焦显微镜实验证实PTGDS与HMOX1存在物理结合。靶向PTGDS(无论是基因沉默还是AT56处理)均可上调HMOX1蛋白水平,并促进其介导的血红素分解,导致细胞内Fe2+水平显著升高。
同时,研究发现PTGDS抑制可上调NCOA4、FTH1、FTL等铁代谢相关蛋白,提示其通过铁蛋白自噬释放储存铁。而铁螯合剂DFO可逆转AT56引起的Fe2+积累、脂质ROS升高及增殖抑制,进一步证明铁代谢紊乱是铁死亡发生的关键环节。更为精巧的是,研究团队构建了HMOX1-H25A突变体,该突变虽不影响其与PTGDS结合,但丧失催化活性,结果发现其无法介导铁死亡相关表型变化,从而确证HMOX1的酶活性在该通路中的必要性。
拓展阅读
铁死亡对血液肿瘤的重要性,核心是突破治疗耐药与提供新方向。血液肿瘤(如AML、淋巴瘤、多发性骨髓瘤)在治疗中常因肿瘤细胞逃避凋亡而产生耐药,而铁死亡作为铁依赖的调控性细胞死亡,可通过脂质过氧化绕开凋亡抵抗,直接破坏肿瘤细胞;多种铁死亡诱导剂(FINs)能与化疗协同增效,部分已进入临床,为复发/耐药患者带来希望。
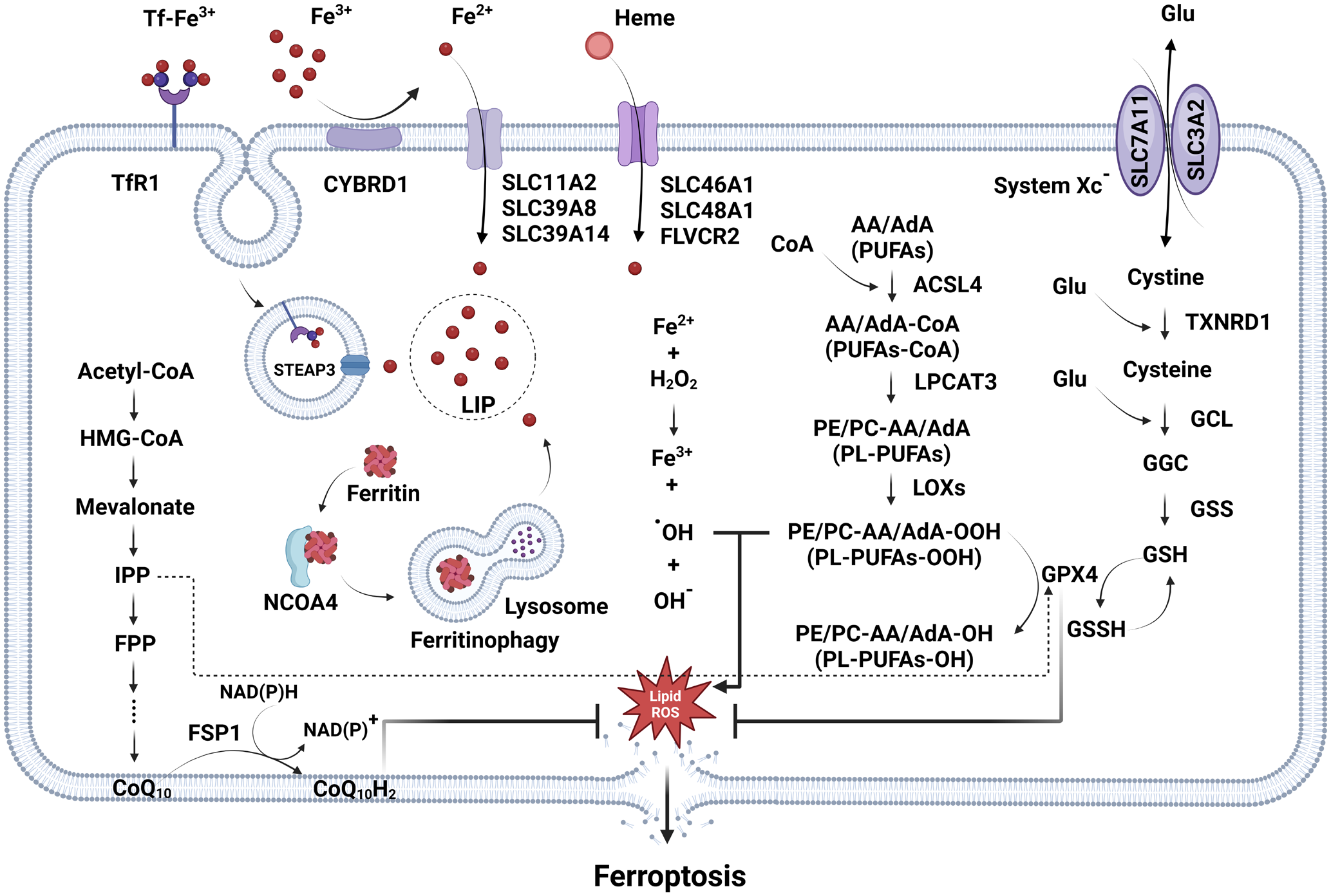
铁死亡作用机制包含三大协同环节:一是抗氧化系统(systemXc--GSH-GPX4轴)的调控,systemXc-由SLC7A11和SLC3A2组成,负责摄取胱氨酸;胱氨酸进入细胞后,经TXNRD1还原为半胱氨酸,半胱氨酸进一步用于合成谷胱甘肽(GSH);GPX4则以GSH为辅酶,将细胞内的脂质过氧化物还原为无毒产物,以此维持肿瘤细胞的氧化还原(redox)平衡;二是铁代谢紊乱的启动,Fe3+通过转铁蛋白(Tf)-转铁蛋白受体1(TfR1)复合物被细胞摄入;摄入的Fe3+经STEAP3还原为Fe2+,随后进入胞内的不稳定铁池(LIP);同时,NCOA4介导的铁蛋白自噬会进一步增加LIP中Fe2+的含量;Fe2+通过Fenton反应生成羟自由基,该自由基可直接攻击细胞膜上的多不饱和脂肪酸(PUFAs);三是脂质过氧化的推进,首先由ACSL4催化PUFAs生成PUFA-CoA,接着LPCAT3将其整合到细胞膜磷脂中,最后LOXs进一步加速磷脂过氧化反应,最终导致细胞膜破裂2。
参考文献:
1.Hu S, et al. Br J Cancer. 2025;132(4):384-400.
2.Shayanmanesh M, et al. J Cell Mol Med. 2025;29(17):e70790.








