




导 语
突触作为神经元间对话的最小单位,在正常脑生理活动中起基础媒介的关键作用,而其损伤变性则会导致各细胞的联系丧失。在阿尔茨海默病(AD)的人类和动物模型中,突触损伤均与认知能力下降密切相关,是AD的主要特征之一。

· 与多种突触蛋白结合或相互作用,产生直接毒性,包括参与突触tau磷酸化、突触蛋白破坏、树突棘丢失等;
· 通过下游钙稳态失调诱导突触毒性(兴奋性毒性和细胞内钙过多对突触功能具有高度破坏性);
· 突触前末端错误折叠的tau蛋白致使突触小泡丢失、耗竭;
· 导致线粒体功能障碍(包括数量减少、结构破坏及线粒体向突触的运输破坏)和随后的自由基生成;
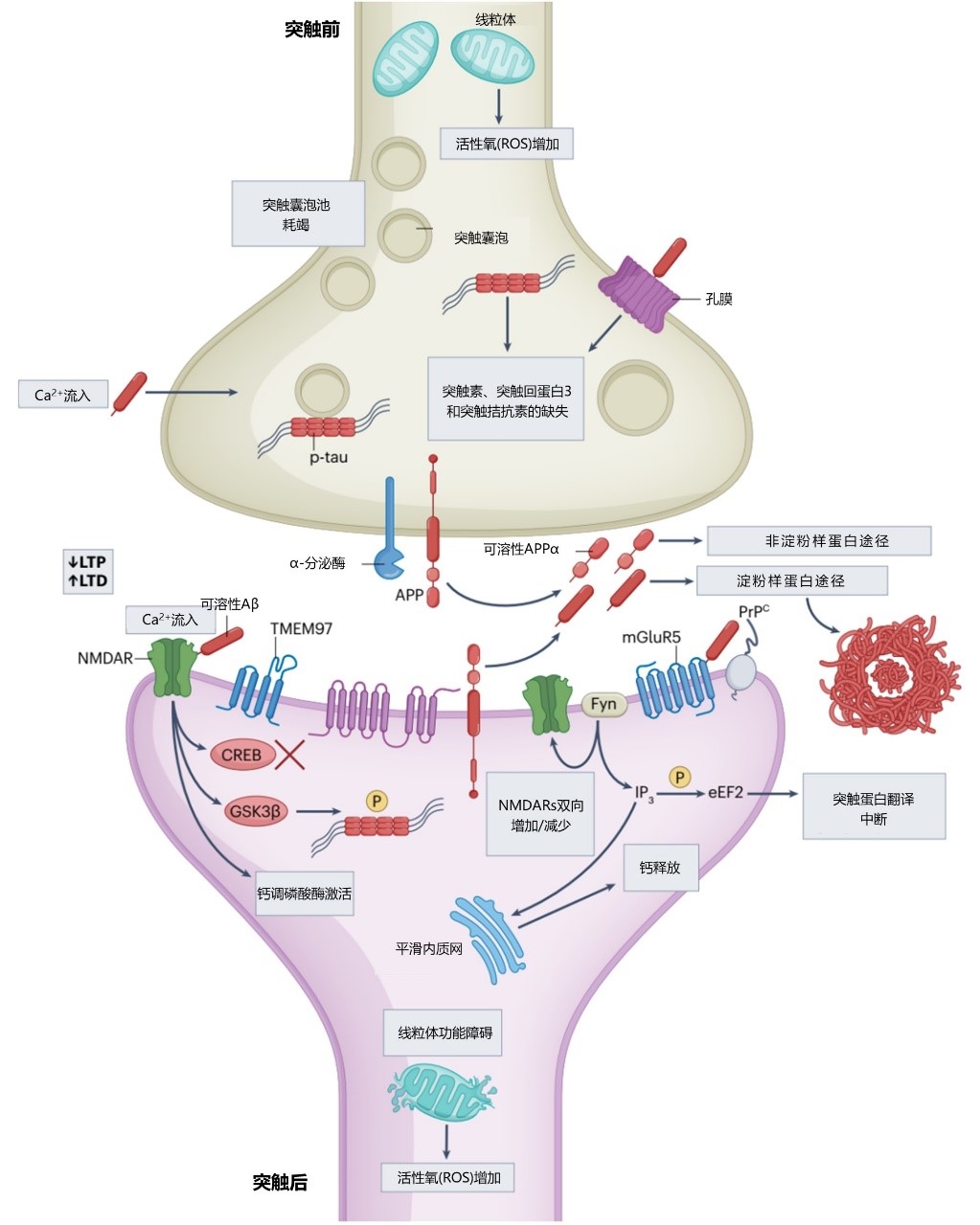
图1. Aβ和tau突触变性在阿尔茨海默病中的推测机制
APP,淀粉样前体蛋白;PrPC,朊蛋白C;mGluR5,代谢型谷氨酸受体 5;eEF2,真核延长因子2;NMDARs,N-甲基-d-天冬氨酸受体;CREB,cAMP 反应组件结合蛋白;LTD,长时程抑制;LTP,长时程增强
传播媒介:突触介导的Aβ和tau的扩散
关键角色:神经胶质细胞的“助纣为虐”
正常生理情况下,小胶质细胞和星型胶质细胞的功能之一,便是修剪大脑发育过程中的冗余突触;然而AD病理(即Aβ 和 tau)及补体系统、炎症细胞因子等多因素,致使功能性突触被异常吞噬(图2)。值得注意的是,AD病理蛋白可通过“标记”突触,使之成为神经胶质细胞清除的目标。基于此,抑制胶质细胞的突触清除,或可改善AD的认知功能障碍。
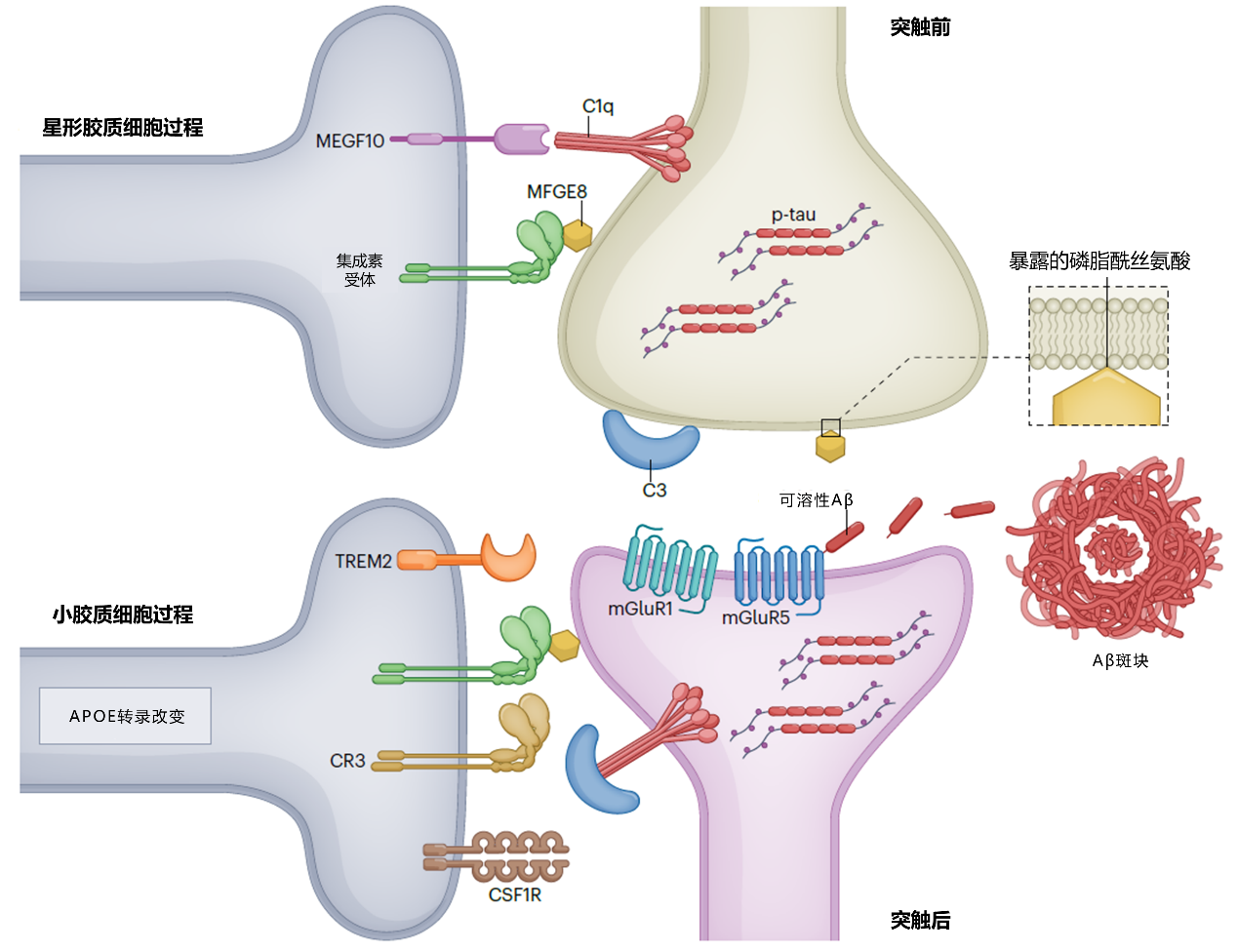
C1q,补体成分1q;C3,补体成分 3;MFGE,乳脂球表皮生长因子;CR3,补体受体 3;CSF1R,集落刺激因子 1 受体;TREM2,髓系细胞触发受体2;mGluR,代谢型谷氨酸受体
觅迹寻踪:突触相关AD生物标志物的探索
突触相关AD生物标志物的开发,目前以突触蛋白为主,其中,大部分研究集中在脑脊液中突触蛋白的检测以及结合突触前囊泡(SV2A)的PET示踪剂的使用:在多项研究中,突触前和突触后蛋白(包括SNAP25、突触受体素、突触素、RAB3A、GAP43 和 AMPA 受体亚基)在 AD 个体的脑脊液水平高于认知正常组对照,是潜在的成像生物标志物(图3);而若未来研究表明,SV2A PET配体在AD个体纵向研究中能准确反映突触丢失,SV2A PET则将成为临床试验中评估治疗保护突触和认知能力的有力工具。
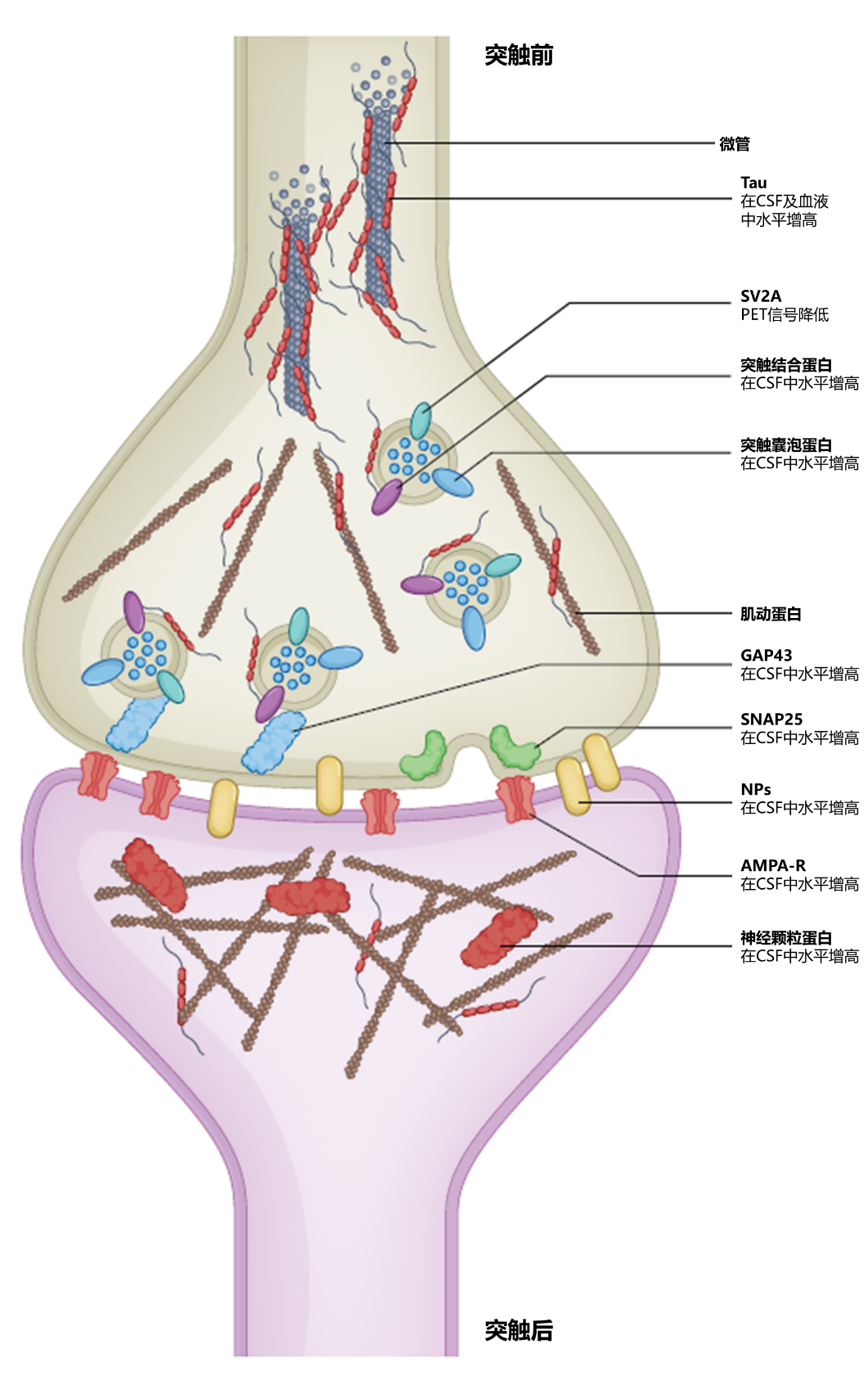
CSF,脑脊液;NPs,神经元正五聚体蛋白;SV2A,突触囊泡糖蛋白 2A;GAP43,生长关联蛋白43;SNAP25,突触体相关蛋白25;AMPA-R,α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体
道阻且长:突触相关靶点探索仍受限
总 结
参考文献:
1. Tzioras M, et al. Synaptic degeneration in Alzheimer disease. Nat Rev Neurol . 2023 Jan;19(1):19-38.
2. Peng L, et al. Mol Psychiatry. The synapse as a treatment avenue for Alzheimer's Disease.2022 Jul;27(7):2940-2949.








